Fragile X syndrome: 80 years since its discovery

The genetic disorder known as fragile X syndrome is the most common known driver of intellectual disabilities worldwide. It occurs when a single gene on the X chromosome ceases to function, thereby failing to code for the protein fragile X messenger ribonucleoprotein 1, or FMRP, which is needed for brain development.
FXS typically becomes evident in by age 2. Children with it display developmental disabilities and social and behavioral problems. Boys are more affected than girls both in terms of number and severity. FXS occurs 1 in 7,000 males and about 1 in 11,000 females, according to the Centers for Disease Control and Prevention. The average lifespan of a person with FXS is normal.
First reported in 1943 by James Purdon Martin and Julia Bell in Britain, the condition was originally referred to as Martin–Bell syndrome. In a biography of Bell, Jesse King explained: “After interviewing affected individuals and detailing the family's history, Martin and Bell suggested that the condition was sex linked, heritable and caused specific sections of the brain to develop improperly.”
In the 80 years since then, researchers have learned a lot about FX, and much of the information now available to patients and caregivers came to be thanks to advocacy organizations.
In 2000, after a prolonged campaign by advocates — in particular the FRAXA Research Foundation — Congress made July 22 officially National Fragile X Awareness Day. In recognition of that effort to spread information and improve outcomes, this article provides an overview of the syndrome, what researchers know about it and still need to determine, and how it’s diagnosed and treated.

The genetics
Almost all cases of the syndrome are caused by an expansion of the trinucleotide repeat CGG within the FMR1 gene on the X chromosome. The extra CGG repeats cause the FMR1 gene to be methylated, which silences it and ultimately leads to reduced expression of the protein product.
The gene in question was first identified in 1991 by researchers in the Netherlands, who published their work in the journal Cell. It varies in length from person to person due to the CGG repeats. Most people have 29 or 30 CGG repeats.
Depending on the number of repeats, three different FMR1 expansions occur:
-
intermediate (45 to 54 CGG repeats)
-
premutation (55 to 200 CGG repeats)
-
full mutation (more than 200 CGG repeats).
It is estimated that 1.5 million people in the U.S. have an FMR1 premutation that is unstable and can expand to full mutation when passed from a mother to her child, causing FXS.
About 100,000 individuals in the U.S. have a full mutation, and men with FMR1 full mutations typically have FXS, whereas only half of the women with full mutations have FXS and the rest either exhibit mild symptoms or are asymptomatic.
Inheritance patterns
FXS is an X-linked dominant condition. Men have one copy of the FMR1 gene, because they have one Y chromosome and one X chromosome, and women have two copies, one on each of their X chromosomes.
A woman with the FMR1 gene has a 50-50 chance to pass it to her children, regardless of their sex. This is because she passes a single copy of the X-chromosome during each pregnancy.
A man with FMR1 full mutations will pass it to all of his daughters but not to his sons, because girls get his X chromosome and boys get his Y chromosome.
People who have an intermediate number of repeats do not have FXS and are not at risk for having children with FXS. However, they might have a slightly higher chance of having some symptoms related to other fragile X-associated disorders and may pass the slightly higher chance of having these disorders to their children.
The inheritance pattern is complicated in premutation cases. People who have 55 to 200 repeats do not have FXS but they might have, or may later develop, other fragile X-associated disorders. In addition, people with a premutation can have children with a premutation or full mutation resulting in FXS.
However, the chances of having a child with a premutation or a full mutation are different for women with a premutation than they are for men with a premutation. A woman with a premutation can pass on a full mutation, whereas a man with a premutation will pass on his permutation — but never the full mutation — to his daughters but not to his sons.
Features of FXS
Every individual with FXS is unique and may exhibit few to many features associated with FXS.
Some of the physical features associated with FXS include large ears, flat feet, seizures, lazy eyes, flexible joints, a long face, a high palate and low muscle tone.
Some of the behavioral features associated with FXS include learning and intellectual disabilities, attention deficit and hyperactivity disorder, anxiety, depression, speech and language delays, autism spectrum disorder, motor delay (late crawling, walking, toileting), and increased sensitivity to sounds, touch, crowds, certain foods and textures.
Children may also exhibit a high tendency to have ear infections.
Diagnosis and treatment
The very first diagnostic genetic test for FXS was done by looking at the chromosomes under a microscope. The name “fragile X” was coined based on the fact that the microscopic examination of revealed a “fragile” X chromosome with a broken end.
Today, however, highly accurate DNA testing is available. Genetic testing includes chromosomal microarray analysis, exome sequencing, and a blood test referred to as “Fragile X CGG repeat analysis” or the “Fragile X DNA test.”
The techniques commonly used are polymerase chain reaction to identify the size of the repetitive section of the FMR1 gene and Southern blot analysis to confirm full mutations and the methylated status of the gene.
The National Fragile X Foundation recommends that any child with intellectual disabilities, developmental delay, speech and language delay, autism or learning disabilities of unknown cause get tested for FXS. Further, it is recommended that any woman with infertility, elevated FSH (follicle-stimulating hormone) levels, premature ovarian failure, primary ovarian insufficiency, or irregular menses get tested.
While there is no cure for FXS, treatments include therapy to learn to talk, walk and interact with others to help people navigate life.
The CDC advises healthcare workers and FXS patients and caregivers to develop treatment and support plans best suitable for individual needs. Taking advantage of the resources available helps individuals with FXS and their families learn new skills and improve their quality of life.
More to know
-
FXS is caused by an expanding mutation that can become bigger when passed on to the next generation. Individuals diagnosed with FXS may lack a family history of FXS.
-
Both girls and boys can have FXS. Boys usually exhibit more severe symptoms, but both boys and girls can exhibit symptoms ranging from normal functioning to severe intellectual disability.
-
Individuals with FXS may have dysmorphic features, but many children do not have any.
-
Diagnosis is important as affected individuals might be helped with therapeutic approaches tailored to individual strengths and weaknesses and behavioral treatment methods.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles
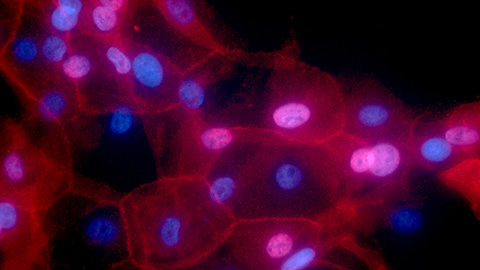
Heat shock proteins as a promising breast cancer therapeutic
Researchers unveiled isoform-specific targets on heat shock protein 90 which may be beneficial in therapeutic development.
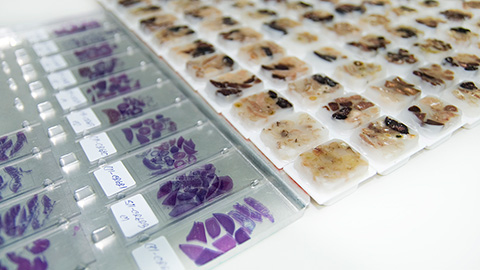
Optimized proteomic analysis of preserved biological tissue samples
Researchers have developed an optimized workflow for analyzing formalin-fixed paraffin-embedded tissue. This workflow provides an enhanced collection of unique proteins and phosphorylation sites for more detailed analysis of biological samples.
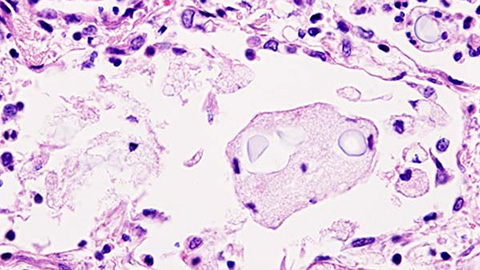
Blood proteomics reveals fungal infection signatures for faster diagnosis
Whole-blood proteomics identifies more than 3,000 host and 160 fungal proteins during cryptococcal infection, offering potential biomarkers for faster diagnosis and improved monitoring without invasive spinal taps.
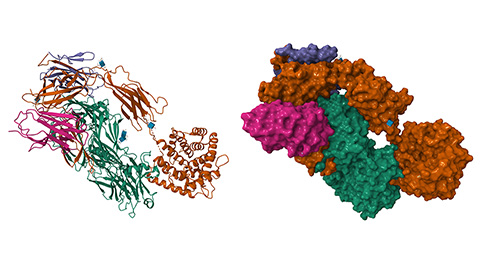
When things get SAPpy: Novel insights into complement
Researchers have defined interactions between an innate immune protein and two of its known binding partners. They identified potential areas of crosstalk between the two binding interactions.
Glutathione pathway implicated in rare disease
Researchers found that glutathione metabolism plays a central role in the pathogenesis of rare disease methylmalonic aciduria using a novel multiomics approach.
A p-value for proteins
Kyoto University researchers developed UniScore, a new tool that uses a target-decoy method to filter false positives in proteomic searches, helping scientists set thresholds and improve reliability when analyzing complex protein data.