‘It’s the DNA. It’s the proteins. And they all are in cahoots.’

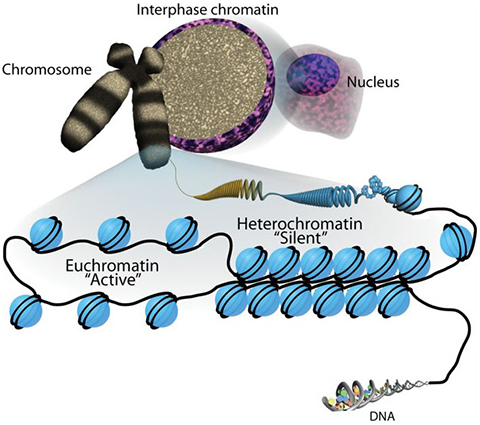
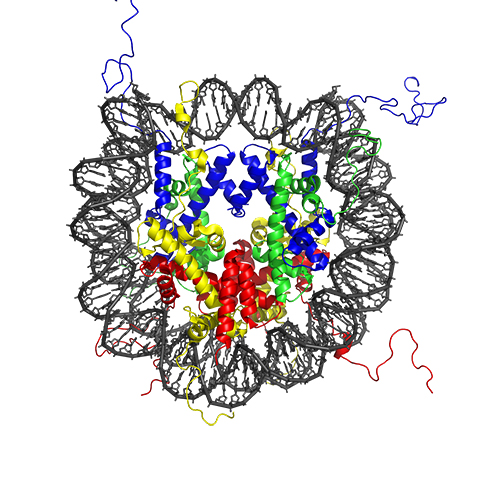
You may have heard that the structure of chromatin resembles a string of beads. The protein beads, or nucleosomes, along a strand of DNA are important in determining the strand’s shape and organization — and they can change over time. DNA is wrapped by the actions of proteins called histones, which form the DNA–histone structures we call nucleosomes. Nucleosomes create a barrier to DNA and our genes. Myriad chromatin-associated machines regulate this barrier by adding or removing histone posttranslational modifications. These PTMs provide the epigenetic information that defines when and how our DNA is accessed.

Brian Strahl, a professor in the biochemistry and biophysics department at the University of North Carolina School of Medicine, is interested in how modifications to nucleosomal histones can alter the structure and activity of DNA.
Histone proteins, and especially their extended tail domains, can pick up a variety of modifications. Each nucleosome is a round cluster of eight histone proteins. Enzymes that add functional groups to and remove them from the amino acids on histone tails have multiple effects: They can make it more or less likely for a gene to be transcribed, mark places where a polymerase or another protein ought to bind, and determine how tightly the genome is compacted. Sometimes, enzymes act in concert, with one modification driving another and producing an additive effect.
ASBMB Today caught up with Strahl to learn more about the histone methyltransferases and other epigenetic enzymes he studies. This interview has been condensed and edited.
How would you describe your research focus?
Our lab thinks a lot about epigenetics, in particular how PTMs work. We are trying to solve how they function to recruit proteins to chromatin and what these interactions mean for downstream events like transcription, replication or repair.
How did you get started in epigenetics?
I got into epigenetics as a grad student looking at transcription. I went to a meeting where for the first time I saw the nucleosome wrapped with DNA. Someone showed a slide of an acetyltransferase acetylating histone tails and the chromatin opening up. I thought, “Wow, that’s what I need to be studying.” Looking at promoter DNA was fun, but I realized that much of gene regulation is dictated by histones and things that modify histones. This was where I wanted to be.
That’s what drew me to Dave Allis’s lab at the University of Virginia. When I was a postdoc with Dave, we put forward this idea of a histone code where different PTMs come together on histones, creating an information messaging system that directs downstream functions in chromatin. Since starting my lab at UNC, I’ve tried to understand the proteins that interact with histones, what those interactions mean and how they work in combination.
You proposed a histone code 20 years ago. Has the idea evolved?
It’s still an unsolved area, and a lot of labs are working on it. There was controversy over whether there’s really a code — some of it was semantics around how you define a code. The histone code probably isn’t as strict as the genetic code; there are patterns built on histones that define what happens downstream, but they may not be completely directive. Rather, they’re going to act in concert with other things like transcriptional regulators, chromatin remodelers and histone variants. They all come together, perhaps more in a chromatin code. We’ve come a long way from those early days, and a main takeaway is that histone PTMs are part of a much larger picture and all of these components are integrated.
Part of the code was also about cross talk, or how modifications on one histone can direct the outcome of others. There’s a lot of evidence still coming out on how these modifications are either repressing or promoting others.
The evidence is pretty strong that there is some kind of information in these modifications. But the honest truth is even the term “epigenetics” is controversial. Some people are very strict that it has to mean information is transferred from mother to daughter cell, whereas others think epigenetics can occur when a gene is up- or downregulated, based on modifications that open or close chromatin but don’t affect the DNA.
So what does epigenetics mean to you?
I take the slightly looser stance. A lot of modifications on histones contribute to the opening of chromatin and transcriptional regulation, but the DNA has not been altered per se. I consider that a form of epigenetic regulation, but I can understand people who want it to be more strictly heritable.
There are some semantics, but I don’t think there’s any doubt now that multiple modifications on histones do function together in a way that can help support, if not direct, certain activities.
What questions are you asking right now in your lab?
We’re asking questions related to how histone methyltransferases function. We’ve been studying several for a long time that methylate histone H3 at lysine 36. One of them is called SETD2; I discovered the yeast homolog Set2 as a postdoc and brought that to my lab. Mammalian SETD2 is an enzyme that associates with RNA polymerase II and travels with it during transcription. We’ve been trying to understand how the enzyme gets to chromatin and its role in transcription.
Set2 and SETD2 led me to other proteins that associate with polymerase and work together to drive methylation, acetylation and even ubiquitylation on histones. And importantly, how these PTMs direct their functions in transcription.
With another methyltransferase, we’ve been interested in its histone reading domains. That’s a theme in chromatin biology: Many of the enzymes that modify histones themselves have domains that bind the same modification they’re producing. We get these read/write mechanisms that help modifications spread along chromosomal arms. Or they sense other modifications: For example, histone acetylation could be a signal for methyltransferase activation. Sometimes these modifications are repulsive and will kick off proteins with the opposite function.
Histone modifications sometimes create chromatin signatures that help other enzymes know where to be: to recognize that landscape and stick around and do something else or to not be there. Often, one modification repels or recruits the production of others and creates a greater PTM landscape, layer upon layer, until we have all kinds of distinct modification types at a region that creates that specific chromatin signature. One example might be the unique set of PTMs found at core promoters — these being lots of H3 and H4 acetylation and certain forms of histone methylation as well as a specific histone variant.
The type of modification signatures getting built could indicate this is a promoter or this is an enhancer, or different modifications might create a signature that we see only in transcribed gene bodies. Our lab has been trying to study how all these modifications become associated and what they do to regulate the chromatin environment that transcription needs.
It seems like a baroque way of labeling DNA.

We have a lot of DNA. We have over 6 billion base pairs, and only 2% of our genome is genes. With all that DNA in the nucleus, we’ve got to ask, How does the cell know where to find a gene? It’s a needle-in-the-haystack kind of problem.
There are layers of organization that begin with a lot of repressed heterochromatin being smushed up against the periphery of the nucleus, while the gene active regions are more interior. Then, within those regions, we have a lot of structure where genes are organized together in little hubs with all the ingredients for gene regulation. Now, within that region where the gene is, how do I know where the promoters are?
A promoter has a unique fingerprint of modifications and even histone variants, and the whole thing says, “Hey, this is where the polymerase should initially bind to get transcription going.” Those regions become harder to find if they don’t have all the modifications there. And, of course, the DNA sequence at promoters is really important to all of this.
What chemical differences might a polymerase complex recognize between an acetylated and a methylated region as it scans through the DNA?
Acetylation neutralizes positively charged lysines. When the histone tails are unacetylated, they tend to collapse and bind to the DNA, so the tails are hidden — which is probably great in the gene bodies but not so great when we want to bring in the polymerase.
When the histone tails become acetylated, the tails pop off the DNA; now they’re more exposed and accessible to build up more transcription modifications. I think this is part of the secret sauce for how modifications may function. As the polymerase moves into the gene bodies, there are other modifications — even DNA methylation. There’s a lot of cross talk.
So we have a combination of histone and DNA modifications at all locations, driving information systems that define promoter enhancer, gene body — or repetitive sequences that we want to keep shut down.
A lot of these modifications not only build a code that directs on and off, but now we’re finding some of them direct chromatin into territories like loops, domains and even liquid–liquid phase-separated droplets that help to bring all these factors together.
It’s an exciting time in the field. We’ve discovered a huge number of distinct modifications and hundreds of places where they exist on histones. But we don’t yet really understand what they all do. Some modifications are more about making the nucleosome more slippery on DNA rather than recruiting an effector protein. We have a lot to learn.
One area that’s been huge in the field is mass spectrometry, which can look at all of the modifications on each individual molecule. Papers on this have found, just in residues 1 to 50 from a histone that’s 115 amino acids long, there are well over 200 distinct combinations of different patterns of modifications in cells.
In less than half of one of the four core histone proteins, you’ve already got 200 possible combinations?
It gets even more complex. In addition to histone PTMs, you have histone variants, DNA, DNA methylation, remodeling machines that use ATP to space the nucleosomes appropriately. All these things are working together.
A little more than 20years ago, we got these exciting ideas about the histone code that I would argue galvanized the field, because no one had really put together that this modification and that modification may work together to recruit a protein with multiple docking domains. That’s been realized; we have multiple domains that read multiple modifications now. But it’s become more complex because it’s not just the histones; it’s all these other things working together. It’s the DNA. It’s the proteins. And they all are in cahoots, all the time. It’s amazing.
I also need to mention that it’s a sad moment in the chromatin field with the recent passing of David Allis. He was such an inspirational person, friend and mentor. He helped the careers of so many — in and outside his lab — and contributed so much to the field. We all miss him dearly.
How did you become a Journal of Biological Chemistry associate editor?
I was on the editorial board for I don’t know how many years — quite a long time. Then all of a sudden, someone nominated me as a potential associate editor. I was just honored. I’ve always felt close to the American Society for Biochemistry and Molecular Biology. I got an ASBMB Young Investigator Award back in 2005, and this has been a home for me. So I’ve always wanted to contribute and be a part of the society.
What’s it like transitioning from the person who writes one review to the person who assesses them all?
It’s really exciting; in looking at all the reviews, you begin to see all the pieces and how they work together to shape a helpful response. My main goal is to help the authors make the best possible paper and to give advice on what is important to focus on for a revision. It’s really fun.

The hardest thing has been when papers have not been reviewed very positively and I have to let people know there are issues or problems. I try to figure out how to relay things in a constructive way — how can they use the comments to develop a better paper to come back with or take somewhere else? A few times, I’ve seen authors take the comments to heart, do a lot of work, and come back with a brand-new paper that is massively revised and way better. And they sailed through review. Those have been really fun experiences. I feel like I’m contributing to science and the passing of information from the lab to the journal.
You were recently an interim chair, you run a lab and you work for the JBC, so a lot of your time is spoken for. But when you aren’t working, what do you like to do?
I play guitar: jazz, blues, some rock. I was in a high school band and in the jazz bands in college. It’s a fun pastime because it can get my mind off work and it takes me someplace else. Sometimes that’s exactly what I need. I wish I had more time to play than I do, but I do enjoy the hobby a lot. My 18-year-old son plays too now, so sometimes we jam out.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in People
People highlights or most popular articles

Huttenhain, Peng win HUPO awards
Huttenhain and Peng received the Distinguished Service Award and Clinical and Translational Proteomics Award, respectively.

Introducing STEM before self-doubt
With hair biology workshops and hands-on STEM programs, Shyretha Brown is building pathways for young girls to see themselves in science. Through Building Bridges, she blends education, identity and access to expand who feels welcome in STEM.

In memoriam: Richard Wolfenden
He was an enzymologist whose work helped spur the development of ACE inhibitor drugs and has been an ASBMB member since 1967.

Tansey named department chair
He has been a faculty member at Otterbein University since 2002.

In memoriam: Joel Habener
He discovered GLP-1, which helped pave the way for transformative diabetes and obesity therapies, and he was an ASBMB member for 25 years.

In memoriam: Walter A. Shaw
He is the namesake for the Walter A. Shaw Young Investigator Award in Lipid Research and founded Avanti Polar Lipids.