JBC: A causal gene for X-linked intellectual disability

Intellectual disability affects between 1 percent and 3 percent of the world’s population. People with X-linked intellectual disability, a heritable condition, present with IQs below 70 and can be mildly or severely handicapped, requiring lifelong care.
The Human Genome Project greatly has facilitated the diagnosis of XLID, and more than 120 genes on the X chromosome have been established as causal for the disease, which primarily affects males.
A recent study in the Journal of Biological Chemistry reports a new causal gene for XLID. The study was conducted by a team of scientists led by Charles Schwartz at the Greenwood Genetic Center in South Carolina and Lance Wells at the University of Georgia.
The family
 Greenwood Genetic Center
Greenwood Genetic Center
The Greenwood Genetic Center serves patients with birth defects, intellectual disability and autism. There, the Schwartz laboratory works with families with histories of XLID and analyzes genetic defects associated with the disease. For the study published in JBC, individuals from the genetics center traveled to Utah to obtain blood samples from a family with members with XLID in order to isolate DNA and generate cell lines.
DNA sequencing of affected family members uncovered many genes with single-nucleotide changes residing on the X chromosome. After looking for variants that resided in the coding region and that resulted in corresponding amino-acid changes, the Schwartz group came across a mutation in an enzyme called O-GlcNAc transferase, or OGT.
The OGT gene variant found in the Utah family had the three characteristics necessary for continued study: It segregated with the disease, contained a single amino-acid change in the protein and was expressed in the brain. That’s when the collaboration with glycobiologist Wells became critical.
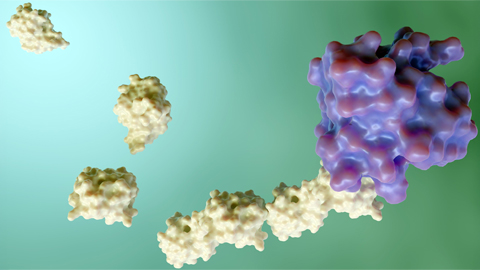
The enzymes
OGT is an enzyme that adds a sugar molecule — GlcNAc — to nuclear and cytosolic proteins. Wells’ group studies this glycosylation modification. The enzyme that removes GlcNAc is called O-GlcNAcase, or OGA. Together, OGT and OGA influence the activity of many cytoplasmic and nuclear proteins in the cell.
The Utah family’s variant contained a leucine-to-phenylalanine substitution, which can be difficult for a protein to accommodate given that phenylalanaine is larger than leucine. The substitution resides in a region of OGT responsible for protein-protein interactions.
Biochemical experiments spearheaded by first author Krithika Vaidyanathan in the Wells laboratory revealed the Utah family’s mutant to be unstable. However, its enzymatic activity was normal, and O-GlcNAc levels, surprisingly, remained constant in patient cell lines derived from blood samples. These findings prompted studies of OGA, the enzyme responsible for removing the modification.
The researchers found that the OGT variant in affected males, along with other co-repressor proteins, occupy the promoter region of the OGA gene and reduce its transcription. This compensates for the OGT variant’s instability and maintains the O-GlcNAc glycosylation modification level.
The Wells laboratory then wondered if the OGT variant occupied other promoter regions too. So the group sequenced RNA of affected males to see which genes’ expression differed from controls’. The researchers found significant changes in several genes, and they’re currently investigating them.
The takeaways
Schwartz notes that the Utah family’s OGT gene variant was challenging to narrow down in the beginning of the work and emphasizes that the collaboration with Wells was critical to establishing OGT’s importance.
Wells points out that the RNA-sequencing experiment involved related patients, which raised the possibility of the results segregating by generation instead of disease. Fortunately, this was not the case.
The researchers say their future studies will investigate additional OGT gene mutations, recently identified by Schwartz and other clinical geneticists, that segregate with the disease. Both the Wells and Schwartz labs are looking at their effects on OGT function. The researchers also are studying stem-cell-derived neuronal cells that are affected by OGT gene mutations.
In the end, the JBC study represents the first instance of an OGT gene mutation being responsible for XLID and broadens the pathways involved in brain development and cognitive function.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Engineering the future with synthetic biology
Learn about the ASBMB 2025 symposium on synthetic biology, featuring applications to better human and environmental health.

Scientists find bacterial ‘Achilles’ heel’ to combat antibiotic resistance
Alejandro Vila, an ASBMB Breakthroughs speaker, discussed his work on metallo-β-lactamase enzymes and their dependence on zinc.

Host vs. pathogen and the molecular arms race
Learn about the ASBMB 2025 symposium on host–pathogen interactions, to be held Sunday, April 13 at 1:50 p.m.

Richard Silverman to speak at ASBMB 2025
Richard Silverman and Melissa Moore are the featured speakers at the ASBMB annual meeting to be held April 12-15 in Chicago.
From the Journals: JBC
How cells recover from stress. Cancer cells need cysteine to proliferate. Method to make small membrane proteins. Read about papers on these topics recently published in the Journal of Biological Chemistry.

ASBMB names 2025 JBC/Tabor Award winners
The six awardees are first authors of outstanding papers published in 2024 in the Journal of Biological Chemistry.