A celebration of our other genome: Mitochondrial DNA

April 25 is National DNA Day, recognizing the 2003 completion of the Human Genome Project. While our nuclear genome gets a lot of attention for good reason, another organelle in our cells has DNA too: the mitochondria. If you get your DNA sequenced with 23andMe, for example, in addition to all your chromosomal ancestry information, you’ll also be told about your mitochondrial DNA origins.
Mitochondria are (in)famously known as “the powerhouse of the cell” because they provide more than 90% of a cell’s energy. Cells use adenosine triphosphate, or ATP, which stores energy in its bonds, like a battery pack to power all their activities. The energy stored in ATP comes from the breakdown of food. While the first step of this process, glycolysis, takes place in the cytosol, the majority of the cell’s ATP is made inside the mitochondria, through aerobic (oxygen-dependent) respiration.
Respiration is also a source of reactive oxygen species, which are both essential components of signaling pathways and harmful sources of oxidative stress leading to aging and cell damage. With a hand in so many critical cellular processes, it’s not surprising that the mitochondria and its DNA are found to have a central role in so many health aspects of our lives, including aging, neurodegeneration and cognitive decline, diabetes, cardiovascular disease, inflammation, exercise capacity and muscular diseases.
Mitochondria are critical in other pathways too, including phospholipid synthesis, thermogenesis, the formation of iron–sulfur clusters that are essential to the structure of many proteins, heme synthesis required for blood to carry oxygen, regulation of calcium signaling (which plays a role in many cell processes), cell death and even influencing epigenetics controlling what nuclear genes are expressed.
Ancient bacteria
All the other organelles in our cells are important too, and they’re all fine with having all their proteins coded for in the nuclear DNA. Why would mitochondria be so special as to have their own DNA? A theory of how mitochondria came to be provides an answer. In 1966 Lynn Margulis proposed the endosymbiotic theory, that mitochondria and chloroplasts are descended from ancient bacteria permanently taken up by other cells, to form the first eukaryotic cells. Other researchers looked down on Margulis and her work at first, so much so that she reportedly received a grant review telling her that her work was so terrible it wasn't worth even attempting to reapply. But she kept going, and by the 1980s, DNA sequencing data was showing that the endosymbiotic theory was realistic. It is now widely accepted as fact.
While mitochondria may have been free living at one point, they can’t survive outside of cells anymore. Mitochondria have been inside cells for two billion years, and one or two things have changed in that time. After the endosymbiotic event, many genes essential to the function of the mitochondria were lost because they were no longer necessary for survival now that mitos were living the good life safe inside another cell. Other genes were lost because there were redundant versions of those genes in the nucleus. Many genes the mitochondria needed that the nucleus didn’t have, were transferred to the nucleus from the mitochondria over time, so the proteins are made outside the mitochondria and imported in. Nowadays, mitochondria contain more than 1,000 different proteins, the vast majority of which are encoded in the nuclear DNA. These include DNA polymerases and repair proteins, all the enzymes for the many metabolic reactions that happen in the mitochondria, and some of the electron transport chain.
The only genes left in mtDNA in humans are a set of genes encoding some essential components of the electron transport chain, as well as a set of tRNAs and ribosomal RNAs necessary for protein synthesis. Just because there aren’t many genes left, doesn’t mean they aren’t important: without these genes, the electron transport chain can’t function, aerobic respiration can’t happen, and many other processes in the mitochondria are disrupted. Without mtDNA, we’d be dead.
mtDNA also appears to encode a few small proteins inside other genes. Inside the 16S ribosomal gene for example, in a sequence translated into a small protein called humanin. This protein has been shown to regulate oxidative stress in some cell types and to play a protective role in Alzheimer’s disease. Not much is known about the significance of these noncanonical mitochondrial genes.
Once mitochondria were happily living as an organelle, why didn’t they give up all their genes to the nucleus? Why keep any DNA? There are probably many reasons. Perhaps evolution is always going on, and the transfer process is not done; eventually all the genes might be transferred to the nucleus, and mitochondria won’t have their own DNA anymore.
Another explanation could be the difficulty of importing proteins with certain characteristics into the mitochondria. To test this hypothesis, researchers have moved genes from the mtDNA to the nucleus, adding a mitochondrial targeting sequence so the protein will be brought to the mitochondria once synthesized. In some cases proteins with hydrophobic regions could not be imported fully into the mitochondria and were degraded instead. These experiments illustrate the idea that one reason mtDNA is preserved may be to prevent having to import these unwieldy proteins. The mitochondria can keep their production in-house thanks to mtDNA.
mtDNA in human health and disease
Most cells can survive without mitochondrial DNA and the corresponding loss of oxidative phosphorylation. After yeast lose their mitochondrial DNA, for example, they struggle for a while, forming tiny slow-growing colonies called “petites.” Then they begin to grow somewhat normally again. They never recover the ability to do oxidative phosphorylation, they rely fully on glycolysis and fermentation, but they adapt and their growth recovers. Most cultured mammalian cell lines can also survive mtDNA loss.
But we humans are more complicated than cells growing in culture, and we cannot live without mtDNA. Our cells, living in our multicellular body with high energy demands and tightly regulated cell processes, need perfectly functioning mitochondria. Any genetic disorder that leads to depletion of mtDNA in humans is lethal before birth, and inherited mutations in mtDNA cause serious diseases including types of neuropathies, epilepsies, stroke disorders, and encephalopathy. mtDNA damage that accumulates during life is associated with cognitive decline, cardiovascular disease, diabetes and many other problems associated with aging.
Mitochondria have many copies of their genome, and there may be many mitochondria per cell. In our bodies, the amount varies greatly. A human cell might have anywhere from hundreds to tens of thousands of mtDNA copies per cell. This is a real contrast to the one perfect copy of nuclear DNA that we generally have per cell. Oocytes, or egg cells, for example have the highest mtDNA copy numbers, in the tens or hundreds of thousands. Cells in the blood are on the lower end in the tens or hundreds. mtDNA can even vary up to ten fold from person to person.
Sometimes though, a variation in mtDNA copy number is a bad sign. As we age, the mtDNA in our blood for example, decreases, with greater decreases being associated with poorer health, including less strength, poorer cognitive abilities, and more likely death from any cause. It’s been suggested that low mtDNA copy number in blood may even be an early biomarker for dementia. There’s some evidence that artificially increasing mtDNA copy number can slow age associated diseases, such as cardiovascular disease, at least in mice.
One consequence of having so many copies per cell is how mtDNA integrity is maintained. In the nucleus, repairing the genome is essential. If a DNA break goes unrepaired, this is enough to kill the cell. The one and only copy of the nuclear genome is precious. In the mitochondria though, DNA repair can be done, but it also has an alternative mechanism to maintain genomic integrity: just throwing away the broken DNA. It has so many copies per mitochondria that throwing out bad copies and expanding the good copies works just as well.
mtDNA in aging and reproduction
Aside from being the powerhouse of the cell mitochondria might be best known for being inherited only from your mother. Once a sperm fertilizes an egg, the mitochondria that were in the sperm are destroyed, leaving only the mitochondria from the egg. This most likely happens for quality control purposes and contributes to the aging reset that happens in babies.
By the time a person reproduces, they’ve been alive for many years, often several decades, and they and their cells have aged, accumulating a fair amount of cellular damage. But babies are somehow born young, even though they come from the cells of an adult. Many things contribute to this age reset, and mitochondrial DNA quality is a factor. During oocyte development there are many layers of quality control mechanisms in place to keep the population of mitochondria and mtDNA the best it can be for the next generation. So by inheriting mitochondria only from its mother, a newly fertilized embryo receives only the best mitochondria. Any less-well-selected mitochondria from the sperm are prevented from messing that up.
That we only inherit mtDNA from our mothers also provides researchers a way to prevent some mitochondrial diseases from being inherited. You’ve probably heard about three-parent babies that receive nuclear DNA from two parents and mtDNA from a donor. This technique allows mothers with mitochondrial diseases to pass on all their genetic information except the mitochondrial DNA.
To do this, doctors take an egg from a donor with healthy mitochondria, and remove the nucleus. Then the nucleus from the mother is removed from her egg, leaving behind the mitochondria, and placed in the donor egg. This new egg can be fertilized and implanted.
The first baby conceived with this technique was born in 2016; the mother’s mtDNA had caused her first two children to die, and the technique allowed her to have a healthy child. When the child’s mtDNA was tested, he appeared to have about 1% of mtDNA from his mother and 99% from the donor. This technique is currently banned in the U.S. because we have rules against genetic modifications that can be passed down to the next generation.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles
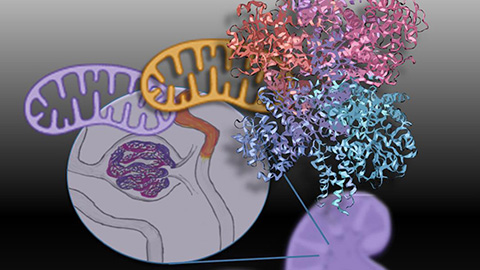
Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.
Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.
Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.
Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.
Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.